


The Food and Drug Administration (FDA) has given its Compliance Program Manual (CPM) for inspecting medical device manufacturers a nip and tuck to align it with requirements found in the US agency’s new Quality Management System Regulation (QMSR).
Implemented on Feb. 2 – the same day the QMSR went into force – the updated CPM 7382.850 lays bare for FDA investigators what they should look and ask for during a facility inspection, among other activities. The CPM effectively replaces the Quality System Inspection Technique, or QSIT, an inspectional approach used by the agency since 1999 and retired on Feb. 1. (Related Story: “R.I.P. QSIT (1999-2026): A Requiem for FDA’s Inspection Technique,” QualityHub, Jan. 27, 2026.)
The FDA will hold a Town Hall on April 1 to give industry insight and advice regarding its updated CPM. (Update: Related Story: “No April Fooling: 3 Must-Know Things From FDA’s Town Hall on Medical Device Inspections,” QualityHub, April 1, 2026.)
In the meantime, QHub Insights asked three consultants and QualityHub subject matter experts – Elisabeth George, Mark Durivage, and Terrence Lowis – to boil down the updated CPM to a handful of key takeaways for device manufacturers to consider as they wait for an FDA investigator to one day conduct a QMSR-based inspection.
Takeaway: Remember, the CPM was written for FDA investigators, not manufacturers.
Nevertheless, “it’s important to be acquainted with this document. It’s great to know what the ‘test’ asks so that you can be prepared,” Elisabeth George said. “If you look at this manual, the FDA is giving you the answers. They’re telling you what the agency thinks is important. They’re telling you why they think it’s important. They’re telling you what they’re looking at and how they’re making some of their decisions. So, it helps us as industry to be better prepared.”
George said it would behoove MedTech quality and regulatory leaders – especially ones newer to the profession – to read not only the current QMSR and its accompanying CPM, but also the former Quality System Regulation (QSR) and the retired QSIT, because they include important information to prep for facility inspections.
She also says understanding Medical Device Single Audit Program (MDSAP) guidelines is useful. The MDSAP program allows a MedTech manufacturer to undergo one quality systems audit to satisfy the regulatory requirements of the US, Canada, Japan, Brazil, and Australia.
The QMSR, the CPM, the QSR, QSIT, and MDSAP “all have ‘niblets’ of information that are invaluable,” George said. “For example, when you’re in an inspection and the FDA starts to go down a path where they’re looking at the risk management style in a design project, and they start asking, ‘Who is involved in this process?’ you’ll understand why, because you’ll understand they’re going down the management responsibility track.”
Takeaway: QMSR inspections are patient- and user-focused based on risk.
While the FDA’s previous QSIT inspectional approach revolved around management, the agency’s retooled CPM decentralizes management and places risk management and patients and users smack-dab in the center of its new Protecting Public Health (PPH) inspectional roadmap. (See figure below; click or tap to expand.)
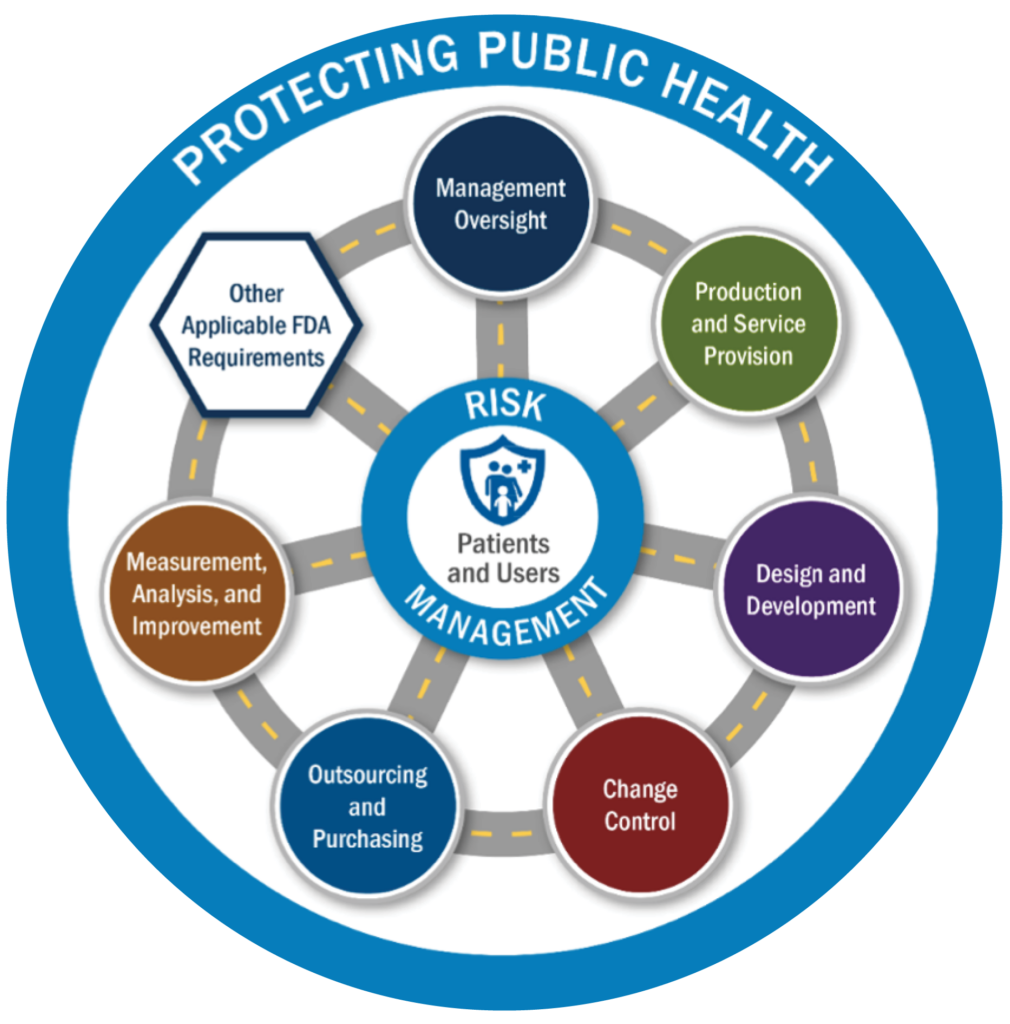
“Patients and users are the central focus of FDA medical device inspections and are depicted at the center,” the agency explains on page 21 of the CPM. “The risk management circle surrounding patients and users represents FDA’s emphasis on using a manufacturer’s risk management documentation to help focus the inspection on risk.”
“The FDA sort of demoted management a little bit, and they’re focusing more on product risk – risk to the patient, risk to the user,” Mark Durivage said. “Now, that’s not to say management is not going to get their feet held to the fire under this CPM. It’s just a little bit different. Everything before with QSIT was focused on what management was doing. But many top managers may not know what a quality policy is and they’re not necessarily involved – although they should be. They care about finances and those sorts of things. So, the FDA is getting smarter about that by focusing more on risk.”
During a facility inspection, Durivage expects investigators to pick areas to focus on based on information they’re seeing and not simply tick off QMSR clauses from a checklist.
“They’re not going to go through things clause by clause by clause by clause by clause. Rather, they’re going to use the PPH model they made based on risk, and they’re going to ‘drive’ to specific areas, and they’re going to look at the elements there,” he said. (Related Brief: “Interpreting FDA’s QMSR Compliance Program: FDA Inspection Types & Corresponding Models,” QualityHub, March 6, 2026.)
From the center of its PPH model, investigators can move to:
- Management Oversight,
- Production and Service Provision,
- Design and Development,
- Change Control,
- Outsourcing and Purchasing,
- Measurement, Analysis, and Improvement, and/or
- Other Applicable FDA Requirements (including adverse events/Medical Device Reports, product recalls, Medical Device Tracking, and Unique Device Identification).
“They’re going to use the data to drive where they go, to see what the issues are and find out what’s causing them,” Durivage said.
Despite the FDA’s longtime recognition of international standard ISO 14971 for risk management, the agency is now “far more embracing of risk management, not just in the total product lifecycle, but in all of the processes,” consultant George said.
“So, manufacturers need to think about their various mechanisms, their tools, their training. Even when they decide how they’re going to train, it’s not so simple,” she said. “People say, ‘Training is training is training.’ But it’s not. Firms need to think about risk-based decision-making, including methods they’re going to use for training. Is it OK to just ‘read and understand’? Or do you need to have a teacher-led program with an assessment tied to it? The bottom line is that it should be a risk-based decision.”
Takeaway: There’s an advantage to knowing old-school QSR language and terms, as well as their new-school QMSR counterparts.
Consultant Durivage said knowing old and new regulatory terms will come in handy when reading and interpreting the FDA’s updated CPM.
“ISO 13485’s ‘Medical Device File’ and [the former QSR’s] ‘Device Master Record,’ or DMR – they’re one and the same. But people are going to have to stop using the acronym DMR. Get it out of your head,” he said.
“A DMR is now a Medical Device File, but for the time being you’re going to see old-school investigators still refer to it as a DMR,” Durivage added. “And you’re going to see new FDA officials, new investigators, call it a Medical Device File because that’s what they’ve been trained to do. So, it doesn’t hurt to be ‘bilingual’ of sorts and know the old and new ways of speaking the regulatory language.”
In a recent QHub Insights article, consultant George pointed out that “it’s OK if [people] accidentally say, for example, ‘Design History Files.’ We’re all human, and it can be difficult to change your vocabulary overnight to say DHF’s replacement, ‘Design and Development Files.’” (Related Story: “From Assessing FDA’s Inspections CPM to Enhancing Supplier Oversight, Here’s 7 Things MedTechs Should Do Right Now,” QualityHub, Feb. 3, 2026.)
Takeaway: Manufacturers need to have strong control over suppliers – and be able to prove it.
Purchasing control activities are heightened under the newly revised CPM, consultant Terrence Lowis warns manufacturers.
“Supplier quality is huge now,” Lowis said. “Investigators will say, ‘If you’re buying things from XYZ, then we want to see everything. What are your quality agreements? What exactly do you stipulate? How often have you audited them?’ It’s all an effort by the FDA to understand if your company is in control of its vendors.”
Under the former QSR, “investigators would say, ‘OK, we read your supplier quality agreement and we read your supplier procedures, and yes, you have all the things that you need.’ But now investigators are saying, ‘I want to see your relationship, I want to see any documentation,’” he said. “And sure, it could be said that the FDA has always done that – but now it’s more of a prescribed, ‘We’re going to walk down a path, and I’m going to look under the hood, because I know exactly what I want to see.’”
“Everything is a deeper dive, and it’s all an effort by the FDA to make sure that all the dots connect.” – Terrence Lowis
This is the FDA’s way of impressing on manufacturers that they must have their ducks in a row when it comes to controlling suppliers.
“FDA investigators are going to go deeper with questions that they would assume would be in your process, whereas before under the old QSR, they would likely just say, ‘All right, you have your process.’ It might not have been very descriptive, that former way, and it left room for a manufacturer to talk their way around not having something or not following something to the T,” Lowis said. “I saw that happen many times over the years.”
When inspecting a company, investigators “are getting more dialed in where they’re now saying, ‘OK, I know you’re hiding behind this clause that you wrote in your supplier quality procedures, but now I need to see more,’” Lowis noted.
“The same goes for complaints; the same goes for your risk management files. Everything is a deeper dive, and it’s all an effort by the FDA to make sure that all the dots connect. The FDA and its investigators are not going to take you at face value that everything is copasetic just because your company’s procedures say you have something or did something.”

